




Содержание
Перейти к:
https://doi.org/2219-5238/2024-32-2-32-41
Перейти к:
Введение. Использование антибиотиков в медицине и ветеринарии привело к их накоплению в природной среде, в том числе в водных объектах питьевого назначения, и формированию устойчивости отдельных видов бактерий к действию антимикробных препаратов. Разработка методик анализа антибиотиков в водных средах актуальна для обеспечения контроля качества питьевой воды на уровне гигиенических нормативов, а также для изучения развития и распространения антибиотикорезистентности.
Цель исследования: разработка методики определения антибиотиков классов макролидов, пенициллинов и фторхинолонов в воде на уровне гигиенических нормативов методом ВЭЖХ/МС-МС.
Материалы и методы. Исследования по разработке методики проведены методом ВЭЖХ/МС-МС на жидкостном хроматографе с масс-спектрометрическим детектором с тройным квадруполем. Извлечение антибиотиков из различных типов проб воды (водопроводной, природной) проводили методом твердофазной экстракции.
Результаты. Разработана селективная и высокочувствительная методика определения 8 антибиотиков в воде. Степень экстракции аналитов из водных матриц составила 72–100 %, диапазон измеряемых концентраций на уровне 0,25–2,50 гигиенических нормативов при анализе проб воды объемом 10 см3, относительная погрешность определения антибиотиков в пробах воды без концентрирования – 20–24 %, с концентрированием на картриджах Oasis HLB – 24–34 %.
Обсуждение. Рассмотрены методические подходы к разработке методики количественного определения антибиотиков групп пенициллинов, макролидов и хинолонов в воде методом ВЭЖХ/МС-МС с применением твердофазной экстракции в качестве пробоподготовки. Полученные результаты согласуются с данными научно-технической и методической литературы. Преимуществом разработанной методики является сокращение времени пробоподготовки, высокая чувствительность в сочетании с анализом незначительного объема образца исследуемой воды.
Заключение. Разработанная методика может быть использована в гигиенических исследованиях содержания остаточных количеств антибиотиков в воде для оценки качества источников питьевого и культурно-бытового водоснабжения.
Нурисламова Т.В., Карнажицкая Т.Д., Старчикова М.О., Терентьев Г.И., Поспелова А.А. Методические подходы к определению антибиотиков в воде на уровне гигиенических нормативов методом высокоэффективной жидкостной хроматографии / масс-спектрометрии. Здоровье населения и среда обитания – ЗНиСО. 2024;32(2):32-41. https://doi.org/2219-5238/2024-32-2-32-41
Nurislamova T.V., Karnazhitskaya T.D., Starchikova M.O., Terentyev G.I., Pospelova A.A. Methodological Approaches to Determination of Antibiotics in Water at the Level of Hygienic Standards Using High-Performance Liquid Chromatography–Mass Spectrometry. Public Health and Life Environment – PH&LE. 2024;32(2):32-41. (In Russ.) https://doi.org/2219-5238/2024-32-2-32-41
Введение. Использование антибиотиков в медицине и ветеринарии привело к их накоплению в природной среде (водоемах, в том числе питьевого назначения, почве, донных отложениях), что явилось одной из причин формирования устойчивости отдельных видов бактерий к действию антимикробных препаратов [1–5]. Важную роль в процессах распространения и сохранения устойчивых патогенов играют природные водные экосистемы, которые служат резервуаром для развития резистентности [6][7]. Устойчивость микроорганизмов к противомикробным препаратам ограничивает перечень антибиотиков для лечения инфекционных заболеваний человека, увеличивает риск развития тяжелых осложнений и распространения инфекций, наносит вред здоровью животных и окружающей среде [8]. Контроль содержания антибактериальных препаратов в питьевой воде и воде водных объектов имеет большое значение для изучения развития и распространения антибиотикорезистентности у возбудителей заболеваний. В рамках реализации Стратегии предупреждения распространения антимикробной резистентности в Российской Федерации на период до 2030 года1 разработаны и внедрены гигиенические нормативы содержания 18 антибактериальных препаратов в водных средах, в том числе в питьевой воде различных источников водоснабжения2 . В настоящее время отечественные методики определения антибиотиков в воде с учетом вновь разработанных гигиенических нормативов в методической базе Российской Федерации отсутствуют.
Исследования содержания антибиотиков в водных средах, проводимые во многих странах мира, выявили присутствие широкого спектра антибиотиков в природных и сточных водах в следовых концентрациях в диапазоне от нг/дм³ до мкг/дм³ [9]. Максимальные концентрации антибиотиков обнаружены в сточных водах фармацевтических предприятий, медицинских учреждений и животноводческих ферм, которые являются основным источником загрязнения природных вод [10]. Приводятся данные об обнаружении антибиотиков в питьевой воде [11].
Определение при совместном присутствии в воде антибиотиков, относящихся к различным структурным группам и обладающих разными физико-химическими свойствами, остается сложной задачей. Для анализа остаточных количеств антибиотиков в водных средах, включая питьевую воду, необходимы высокочувствительные и селективные методики. В настоящее время перспективным методом анализа низких концентраций антибиотиков в водных средах, продуктах питания, почве и других объектах является метод высокоэффективной жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (ВЭЖХ/МС-МС) из-за высокой чувствительности и высокой селективности детектора [12]. Особое внимание при разработке методик количественного анализа антибиотиков в водных средах уделяется отработке условий извлечения аналитов из матрицы. Одним из эффективных и простых в техническом плане способов извлечения целевых компонентов из водных сред является твердофазная экстракция (ТФЭ), включающая селективное извлечение, очистку экстракта при необходимости и концентрирование пробы за одну процедуру [13–15]. Для эффективного извлечения антибиотиков из водных сред методом ТФЭ успешно применяются картриджи Oasis HLB, способные удерживать широкий спектр полярных и неполярных молекул с кислотными, нейтральными и основными свойствами [11][16–19].
Цель исследования: разработка методики определения антибиотиков классов макролидов, пенициллинов и фторхинолонов в воде на уровне гигиенических нормативов методом ВЭЖХ/МС-МС.
Материалы и методы. Исследования по разработке методики определения 8 антибиотиков (азитромицина, амоксициллина, ампициллина, бензилпенициллина, кларитромицина, оксациллина, ципрофлоксацина и эритромицина) в воде проведены в период 2022–2023 гг. на жидкостном хроматографе с масс-спектрометрическим детектором LCMS-8050 с тройным квадруполем (Shimadzu, Япония). Стандартные растворы антибиотиков для построения градуировочных кривых готовили растворением аналитических стандартов с массовой долей основного вещества 93,8–99,5 % (производство Merck, EDQM, Dr. Ehrenstorfer Reference Materials) в метаноле с последующим разбавлением деионизированной водой до нужной концентрации.
Масс-спектрометрическое детектирование проводили с использованием электроспрея в режиме генерации положительно заряженных ионов. Параметры масс-спектрометрического детектора LC/MS 8050, отвечающие за поступление целевых компонентов из жидкостного хроматографа в масс-спектрометрический детектор, представлены в табл. 1.
Таблица 1. Параметры масс-спектрометрического детектора LC/MS 8050
в режиме тонкой настройки
Table 1. Parameters of the ion detector of LC/MS 8050 in fine tuning mode
|
Показатель / Indicator |
Параметры / Parameters |
|
Ионизация / Ionization |
Электростатическим распылением (ESI) |
|
Полярность / Polarity |
Положительная / Positive |
|
Поток газа-осушителя (азота) / Drying gas flow (nitrogen) |
5 л/мин / L/min |
|
Поток газа-распылителя (азота) / Nebulizer gas flow (nitrogen) |
2 л/мин / L/min |
|
Поток нагревающего газа (воздуха) / Heating gas (air) flow |
5 л/мин / L/min |
|
Температура интерфейса / Interface temperature |
300 °С |
|
Температура нагревательного блока / Heating block temperature |
400 °С |
|
Температура линии десольватации / Desolvation line temperature |
250 °С |
|
Напряжение на интерфейсе / Interface voltage |
4,0 kV |
|
Сила тока на интерфейсе / Interface current |
0,4 мкА / μA |
Определение родительских и дочерних ионов антибиотиков проводили с использованием программного обеспечения LabSolution (Shimadzu, Япония) в автоматическом режиме, анализируя стандартные растворы исследуемых соединений с концентрациями 100 нг/см³. Тип сканирования – мониторинг множественных реакций (MRM). В качестве основных дочерних ионов для каждого антибиотика выбирали дочерний ион, имеющий максимальный отклик сигнала детектора. Остальные дочерние ионы, полученные при фрагментации индивидуального антибиотика, отнесены к подтверждающим дочерним ионам.
Изучение эффективности хроматографического разделения 8 антибиотиков проводили на колонке с обращенно-фазным сорбентом ZORBAX Eclipse XDB-C18 длиной 150 мм, внутренним диаметром 2,1 мм зернением 3,5 мкм (Agilent Technologies, США) с использованием стандартных растворов изучаемых соединений в метаноле с концентрациями 100 нг/см³. Экспериментально подбирали количественный состав подвижной фазы, состоящий из смеси метанола и деионизированной воды с добавлением в качестве модификатора муравьиной кислоты с концентрацией 0,02; 0,05; 0,1; 0,2 %, и режим градиентного элюирования. Для оценки хроматографического разделения антибиотиков рассчитывали коэффициент селективности α (фактор разделения) и разрешение пиков R (степень разделения)3. Разделение считается полным при условии α ≥ 1,0; R ≥ 1,0.
Отработку условий извлечения антибиотиков способом твердофазной экстракции (ТФЭ) проводили на образцах питьевой воды централизованного водоснабжения, природной воды поверхностных и подземных источников и воды бассейнов. Изучали эффективность применения картриджей Oasis HLB (сорбционная емкость 60, 200 и 500 мг) и картриджей Strata C18-E (сорбционная емкость 500 мг). Перед проведением процедуры ТФЭ картриджи кондиционировали, последовательно пропуская через них 5 см³ органического растворителя и 5 см³ деионизированной воды. Пропускали через подготовленные картриджи водные стандартные растворы объемом 10–100 см³ с известной концентрацией антибиотиков самотеком, без применения вакуума, промывали 2 см³ деионизированной воды и элюировали с картриджа различными объемами ацетонитрила или метанола в условиях кислой и нейтральной среды. Полученный экстракт концентрировали в 10–50 раз в токе воздуха и анализировали методом ВЭЖХ/МС-МС. Наличие «проскока» при проведении ТФЭ устанавливали путем анализа целевых компонентов в сливах после нанесения стандартных образцов на картриджи и промывки сорбента после нанесения образцов. Степень экстракции (R) рассчитывали по формуле4:
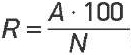 ,
,
где
R – степень экстракции вещества, %;
А – количество вещества, извлеченное органическим растворителем;
N – заданное количество вещества в водном образце.
Градуировочные характеристики, выражающие зависимость сигнала детектора от массовой концентрации основного дочернего иона антибиотика в анализируемом растворе устанавливали методом абсолютной градуировки с проведением твердофазной экстракции перед хроматографическим анализом за исключением бензилпенициллина, оксациллина, ампициллина (по причине высоких ПДК анализ данных веществ не требовал концентрирования и дополнительных этапов пробоподготовки). Диапазон измеряемых концентраций антибиотиков в водных средах подбирали с учетом предельно допустимых концентраций и ориентировочных допустимых уровней в соответствии с СанПиН 1.2.3685–21 (табл. 5) с кратностью от 0,25 до 2,5 относительно гигиенического норматива.
Исследования по оценке погрешности выполнения измерений концентраций антибиотиков в воде проводили методом «задано – получено» с использованием в качестве матриц питьевой воды централизованного водоснабжения, природной родниковой воды и воды бассейна. На основании экспериментальных данных рассчитывали метрологические показатели методики определения массовых концентраций 8 антибиотиков в водных средах методом ВЭЖХ/МС-МС, характеризующие степень достоверности результатов измерений.
Апробацию методики проводили на образцах водопроводной воды систем централизованного водоснабжения, природной воды и воды плавательных бассейнов, отобранных в осенний период 2023 г.
Результаты. Проведена оптимизация параметров масс-спектрометрического детектора в режиме мониторинга множественных реакций (МРМ) для идентификации антибиотиков, в результате которой зарегистрированы родительские и дочерние ионы в условиях оптимального соотношения между напряжением фрагментора и энергией ячейки соударения (табл. 2). В качестве основного дочернего иона для каждого антибиотика выбран ион, имеющий максимальный отклик сигнала детектора (100 %) (табл. 2), который использовался для количественного расчета. Остальные дочерние ионы, полученные при фрагментации индивидуальных соединений, отнесены к подтверждающим дочерним ионам.
Таблица 2. Родительские и дочерние ионы, полученные в результате оптимизации
параметров масс-спектрометрического детектора
в режиме тонкой настройки для идентификации антибиотиков
Table 2. Parent and daughter ions obtained as a result of optimization
of parameters of a mass spectrometric detector
in fine-tuning mode for identification of antibiotics
|
Наименование вещества / Name of substance |
Брутто формула / |
Мол. масса, М / |
Родительский ион [МН+], m/z / |
Дочерние ионы, m/z / |
Интенсивность сигнала дочерних ионов, % / |
|
Амоксициллин / Amoxicillin |
C16H19N3O5S |
365,4 |
366,1 |
114,10 |
100 |
|
208,10 |
35,7–66,3 |
||||
|
Ампициллин / Ampicillin |
C16H19N3O4S |
349,406 |
350,1 |
106,10 |
100 |
|
191,90 |
21,0–39,0 |
||||
|
160,10 |
17,5–32,5 |
||||
|
Азитромицин / Azithromycin |
C38H72N2O12 |
748,99 |
749,2 |
591,20 |
100 |
|
158,05 |
39,9–74,1 |
||||
|
116,10 |
37,8–70,2 |
||||
|
Эритромицин / Erythromycin |
C37H67NO13 |
733,939 |
734,2 |
158,00 |
100 |
|
576,10 |
39,2–72,8 |
||||
|
Оксациллин / Oxacillin |
C19H19N3O5S |
401,44 |
402,1 |
160,10 |
100 % |
|
242,90 |
71,4–132,6 |
||||
|
Бензилпенициллин / Benzylpenicillin |
C16H18N2O4S |
334,39 |
334,90 |
159,90 |
100 |
|
176,00 |
69,3–128,7 |
||||
|
Кларитромицин / Clarithromycin |
C38H69NO13 |
747,96 |
748,2 |
158,00 |
100 |
|
590,10 |
32,2–59,8 |
||||
|
Ципрофлоксацин / Ciprofloxacin |
C17H18FN3O3 |
331,347 |
332,1 |
230,90 |
100 % |
|
288,10 |
37,1–68,9 |
Примечание: жирным шрифтом выделены основные дочерние ионы.
Note: Major daughter ions are shown in old.
Изучение условий хроматографического разделения 8 антибиотиков показало качественное разделение амоксициллина, ципрофлоксацина ампициллина, азитромицина на колонке ZORBAX Eclipse XDB-C18 (2,1 × 150 мм) зернением 3,5 мкм в условиях градиентной подачи подвижной фазы, состоящей из смеси метанола и 0,1 % раствора муравьиной кислоты в деионизированной воде со скоростью 0,3 см³/мин (табл. 3). Идентификацию эритромицина, бензилпенициллина, оксациллина и кларитромицина проводили по наличию на хроматограмме характерных родительского и дочерних ионов.
Таблица 3. Режим градиентной подачи подвижной фазы
для эффективного разделения анализируемых компонентов
Table 3. The HPLC gradient mode of mobile phase
for efficient separation of analyzed components
|
Время, мин / Time, min |
Объемная доля метанола (В), % / |
Объемная доля 0,1 % водного раствора муравьиной кислоты (А), % / |
|
0,00–1,00 |
0 |
100 |
|
1,00–5,00 |
0–50 |
100–50 |
|
5,00–5,01 |
50–65 |
50–35 |
|
5,01–8,00 |
65 |
35 |
|
8,00–8,01 |
65–100 |
35–0 |
|
8,01–11,00 |
100 |
0 |
|
11,00–11,01 |
100–0 |
0–100 |
|
11,01–14,00 |
0 |
100 |
Значения параметров хроматографического разделения изучаемых антибиотиков в подобранных условиях анализа представлены в табл. 4. Коэффициенты селективности (α) составили 1,0–1,2, значения коэффициентов разрешения пиков (R) – больше 1, за исключением бензилпенициллина и кларитромицина.
Таблица 4. Режим градиентной подачи подвижной фазы
для эффективного разделения анализируемых компонентов
Table 4. The HPLC gradient mode of mobile phase
for efficient separation of analyzed components
|
Наименование вещества / |
Время выхода пика, мин / |
Коэффициент селективности, α / |
Коэффициент разрешения пиков, R / |
Ширина пиков W, мин / |
|
Амоксициллин / Amoxicillin |
4,852 |
>1 |
– |
0,1 |
|
Ципрофлоксацин / Ciprofloxacin |
6,008 |
1,2 |
11,6 |
0,1 |
|
Ампициллин / Ampicillin |
6,27 |
1,0 |
2,6 |
0,1 |
|
Азитромицин / Azithromycin |
6,803 |
1,1 |
4,3 |
0,15 |
|
Эритромицин / Erythromycin |
7,576 |
1,1 |
6,2 |
0,1 |
|
Бензилпенициллин / Benzylpenicillin |
7,757 |
1,0 |
0,4 |
0,16 |
|
Оксациллин / Oxacillin |
8,209 |
1,1 |
2,5 |
0,2 |
|
Кларитромицин / Clarithromycin |
8,247 |
1,0 |
0,3 |
0,1 |
Хроматограмма стандартной смеси азитромицина, амоксициллина, ампициллина, бензилпенициллина, кларитромицина, оксациллина, ципрофлоксацина и эритромицина в метаноле с концентрациями 100 нг/см³ представлена на рис. 1.
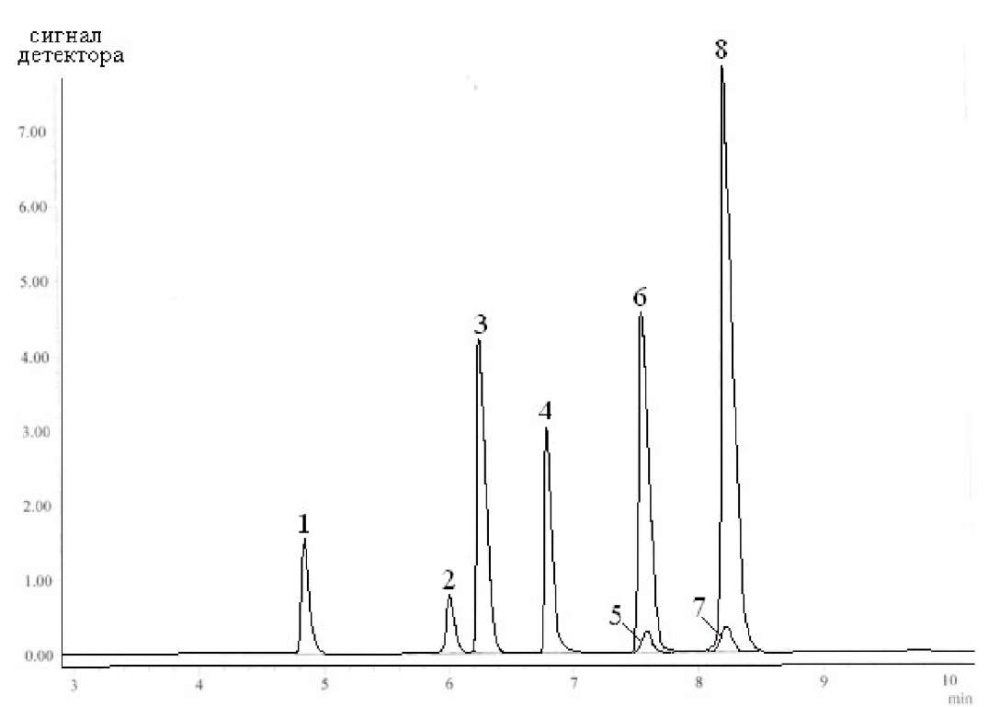
Рис. 1. Хроматограмма разделения стандартной смеси антибиотиков
на колонке ZORBAX Eclipse XDB-C18 в градиентном режиме элюирования.
Время выхода: 1 – амоксициллин, 2 – ципрофлоксацин, 3 – ампициллин,
4 – азитромицин, 5 – эритромицин, 6 – бензилпенициллин,
7 – оксациллин, 8 – кларитромицин
Fig. 1. Chromatogram of separation of a standard mixture of antibiotics
on a ZORBAX Eclipse XDB-C18 column in gradient elution mode.
Retention time: 1 – amoxicillin (4.85 min), 2 – ciprofloxacin, 3 – ampicillin,
4 – azithromycin, 5 – erythromycin, 6 – benzylpenicillin,
7 – oxacillin, 8 – clarithromycin
Изучение эффективности извлечения антибиотиков из водных сред способом твердофазной экстракции показало, что применение классического обращенно-фазного сорбента С18 на основе силикагеля (Strata C18-E) не позволило добиться удерживания амоксициллина на картридже Strata, при нанесении стандартного образца объемом 10 см³ потери аналита составили 45 %.
При отработке условий проведения твердофазной экстракции с использованием картриджей Oasis HLB установлено, что использование патронов с сорбционной емкостью 60 мг (Oasis HLB 3СС/60 мг) приводит к большим потерям амоксициллина (до 85 %) и ампициллина (до 25 %) на этапе нанесения пробы объемом 10 см³ на картридж. Отсутствие «проскоков» аналитов наблюдалось на картриджах с большей сорбционной емкостью – Oasis HLB 6СС/500 мг при загрузке стандартных водных растворов антибиотиков объемом 10 см³ без применения вакуума или избыточного давления. Данный объем пробы достаточен для анализа изучаемых соединений с чувствительностью на уровне 0,25–2,5 гигиенических нормативов и нижним пределом определения изучаемых антибиотиков в воде 5–20 нг/дм³ с учетом концентрирования полученных экстрактов в 50–10 раз. Увеличение объема пробы до 100 см³ приводило к существенному повышению потери аналитов на этапе нанесения пробы на картридж. Экспериментально установлено, что для ципрофлоксацина потери составляли до 90 %, азитромицина – до 10 %.
В зависимости от различных кислотно-основных свойств изучаемых аналитов, величин ПДК, отличающихся более чем в 1000 раз, и чувствительности детектора отработаны два варианта подготовки проб воды к анализу с использованием картриджей Oasis HLB 6СС/500 мг – в условиях кислой и нейтральной среды. Вариант 1 – для извлечения эритромицина из водных сред предварительно промывали картридж 5 см³ метанола и 5 см³ деионизированной воды, загружали на картридж 10 см³ пробы воды, промывали 2 см³ деионизированной воды и элюировали эритромицин 10 см³ метанола. Для достижения необходимой чувствительности полученный экстракт концентрировали в 10 раз при плюс 40 °С в токе воздуха, фильтровали через фильтр с размером пор 0,2 мкм и анализировали в подобранных хроматографических условиях. Вариант 2 – твердофазную экстракцию азитромицина, амоксициллина, кларитромицина и ципрофлоксацина из проб воды проводили, предварительно пропуская через картридж 5 см³ 0,1 % раствора муравьиной кислоты в метаноле, 5 см³ деионизированной воды, загружали на картридж 10 см³ пробы воды, промывали 2 см³ деионизированной воды и элюировали целевые компоненты 5 см³ 0,1 % раствора муравьиной кислоты в метаноле. Полученный экстракт концентрировали в 50 раз при плюс 40 °С в токе воздуха, фильтровали через фильтр с размером пор 0,2 мкм и анализировали в подобранных хроматографических условиях. Время проведения ТФЭ одной пробы не превышает 15 минут, при использовании устройства для твердофазной экстракции в течение 15 минут можно провести одновременную экстракцию до 10 проб.
Исследование возможности применения процедуры высушивания экстракта досуха после ТФЭ показало, что в случае изучаемых антибиотиков (азитромицина, амоксициллина, кларитромицина и ципрофлоксацина) недопустимо полное упаривание экстракта из-за больших потерь целевых соединений. В ходе экспериментов установлено, что при полном высушивании экстрактов при комнатной температуре потеря ципрофлоксацина и эритромицина достигала 25 %, азитромицина и амоксициллина – 10 %. С целью снижения потери анализируемых соединений при высушивании экстракта в токе воздуха на первом этапе упаривали пробу до 1 см³, к концентрату добавляли 0,1 см³ деионизированной воды, перемешивали и затем упаривали пробу до объема 0,2 см³. Проведение ТФЭ в приведенных условиях обеспечивает извлечение ципрофлоксацина на уровне 73 %, кларитромицина – 100 %, азитромицина – 72 %, амоксициллина – 89 %, эритромицина – 100 %. Определение антибиотиков пенициллинов (ампициллина, бензилпенициллина и оксациллина) проводили без концентрирования проб, так как чувствительность масс-спектрометрического детектора при прямом вводе проб воды достаточна для их определения на уровне 0,25–2,5 ПДК. Анализируемые пробы воды предварительно фильтровали через целлюлозно-ацетатный фильтр с размером пор 0,2 мкм, отбрасывая первые 5 см³ фильтрата, и анализировали в подобранных хроматографических условиях.
Градуировочные зависимости y = ax + b для определения антибиотиков в анализируемом растворе представлены на рис. 2.
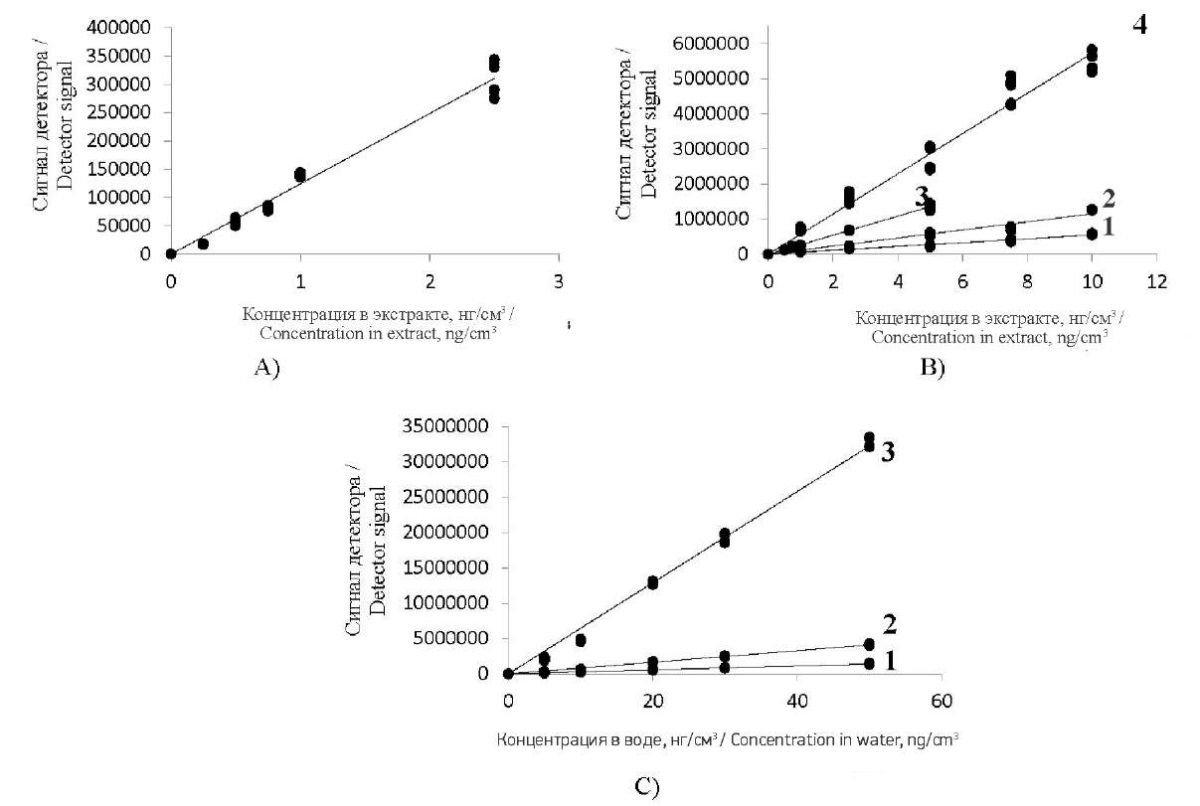
Рис. 2. Градуировочные графики для определения (А): азитромицина;
(B): ципрофлоксацина (1), амоксициллина (2), эритромицина (3), кларитромицина (4);
(C): ампициллина (1), бензилпенициллина (2), оксациллина (3)
Fig. 2. Calibration curves for the determination of (A): azithromycin;
(B): ciprofloxacin (1), amoxicillin (2), erythromycin (3), clarithromycin (4);
(C): ampicillin (1), benzylpenicillin (2), oxacillin (3)
Градуировочные коэффициенты (К) для измерения массовых концентраций антибиотиков в водных средах составили 1,7 × 10–7 … 4,7 × 10–5. Достоверность аппроксимаций (R²) варьировала в пределах 0,9612–0,9937, погрешность построения градуировочных зависимостей составила от 11,5 % для ампициллина до 14,5 % для ципрофлоксацина. Нижний предел обнаружения (НПО) антибиотиков составил 5–20 нг/дм³. Диапазоны измеряемых концентраций в воде, гигиенические нормативы содержания и класс опасности изучаемых антибиотиков представлены в табл. 5. В заданных диапазонах измерений сохраняется линейность зависимости сигнала масс-спектрометрического детектора от концентрации основного дочернего иона определяемого антибиотика в анализируемом растворе.
Таблица 5. Наименование антибиотиков, диапазоны измерения,
предельно допустимые концентрации (ПДК)
и ориентировочные допустимые уровни (ОДУ) в воде, класс опасности
Table 5. Name of antibiotics, measurement ranges, maximum permissible concentrations (MPC)
and approximate permissible levels (APL) in water, hazard class
|
Наименование вещества / |
Диапазон измерения, мг/дм³ / |
ПДК (ОДУ) в воде, мг/дм³ / |
Класс опасности / |
|
Азитромицин / Azithromycin |
0,000005–0,00005 |
0,000019 |
1 |
|
Амоксициллин / Amoxicillin |
0,00002–0,0002 |
0,000078 |
1 |
|
Кларитромицин / Clarithromycin |
0,00002–0,0002 |
0,00012 |
1 |
|
Ципрофлоксацин / Ciprofloxacin |
0,00002–0,0002 |
0,000089 |
1 |
|
Эритромицин / Erythromycin |
0,00005–0,0005 |
0,0002 |
1 |
|
Ампициллин / Ampicillin |
0,005–0,05 |
0,02 |
2 |
|
Бензилпенициллин / Benzylpenicillin |
0,005–0,05 |
0,02 |
2 |
|
Оксациллин / Oxacillin |
0,005–0,05 |
0,02 |
2 |
Проведена метрологическая аттестация методики выполнения измерений массовых концентраций азитромицина, амоксициллина, ампициллина, бензилпенициллина, кларитромицина, оксациллина, ципрофлоксацина и эритромицина в воде методом ВЭЖХ/МС-МС. Показатель точности (границы относительной погрешности при доверительной вероятности Р = 0,95) определения азитромицина в воде составляет 34 %, амоксициллина – 32 %, кларитромицина – 29 %, ципрофлоксацина – 33 %, оксациллина – 24 %, бензилпенициллина – 20 %, ампициллина – 22 %, эритромицина – 24 %. Показатели повторяемости и воспроизводимости определены в диапазонах 6–14 и 7–16 % соответственно.
Проведены исследования содержания антибиотиков в образцах воды, отобранных в летний период на территории крупного промышленного центра: точка 1 – вода питьевая централизованного водоснабжения, точка 2 – ключевая вода в черте города, точка 3 – вода плавательного бассейна, точка 4 – ручей, расположенный рядом с лечебным учреждением (стационар), точка 5 – речная вода, точка 6 – речная вода в точке сброса очищенных бытовых сточных вод. Анализ проводился в день отбора проб на жидкостном хроматографе с масс-спектрометрическим детектором с тройным квадруполем LSMS 8050 (Shimadzu). В результате анализа отобранных образцов воды не обнаружено содержание антибиотиков ни в одной пробе на
уровне выше НПО, равного 0,25 ПДК для каждого изучаемого антибиотика. Отрицательные результаты содержания антибиотиков в пробах воды, возможно, связаны с процессами фотодеградации, сорбцией донными отложениями, протекающими в водоемах, или отсутствием источников выделения изучаемых антибиотиков на территории обследования.
Обсуждение. Рассмотрены методические подходы к разработке методики количественного определения антибиотиков групп пенициллинов, макролидов и хинолонов в воде методом ВЭЖХ/МС-МС с применением твердофазной экстракции в качестве пробоподготовки. Полученные результаты согласуются с данными научно-технической и методической литературы [20–26].
Применение метода тандемной хромато- масс-спектрометрии обеспечило возможность селективного обнаружения антибиотиков по характерным масс-спектрам родительских и дочерних (основных и подтверждающих) ионов, установленные значения которых соответствуют опубликованным данным [20].
Примененный метод ТФЭ для подготовки образцов воды к анализу показал высокую степень извлечения изучаемых антибиотиков. Так, степень экстракции эритромицина на липофильно-гидрофильном картридже Oasis HLB в отработанных нами условиях составляет 100 %, амоксициллина – 89 %, в то время как экстракция на обращенно-фазовом картридже Strata X из проб большего объема не превышает 37,2 % [21] и 33 % [22] соответственно. Степень экстракции остальных антибиотиков установлена на уровне 72–100 %.
Нижние пределы определения (НПО) антибиотиков в образцах воды объемом 10 см³ составили 5–20 нг/дм³. Низкие значения НПО сопоставимы с результатами хромато-масс-спектрометрических исследований тех же классов антибиотиков с концентрированием на картриджах Oasis HLB в образцах воды большего объема от 500 см³ [18][23] до 1000 см³ 5 [24][25]. Повышение чувствительности определения антибиотиков на фоне анализа небольшого объема образца, возможно, связано со снижением влияния матричного эффекта, зависимого в том числе от кратности концентрирования анализируемой пробы воды.
Погрешность количественного определения антибиотиков в воде методом абсолютной градуировки с проведением процедуры ТФЭ составила 24–34 %, при прямом вводе проб – 20–24 %. Значения погрешности и ее составляющих сопоставимы с уровнями погрешностей, полученными при использовании метода внутреннего стандарта для построения градуировочной характеристики и количественного хромато-масс-спектрометрического анализа антибиотиков классов макролидов, пенициллинов и фторхинолонов в водных средах [26].
Преимущество разработанной методики заключается в существенном сокращении времени подготовки проб и в целом продолжительности одного анализа, не превышающей 30 мин.
Апробация методики проведена при исследовании проб воды источников централизованного водоснабжения, природной воды и воды плавательных бассейнов. Результаты исследований показали отсутствие изучаемых антибиотиков в проанализированных пробах в концентрациях на уровне и выше 0,25 ПДК и 0,25 ОДУ, что, вероятно, может быть связано с отсутствием источников выделения изучаемых антибиотиков на территории обследования или другими, не изученными в настоящей работе факторами.
Ограничения исследования. Связаны с ограниченным по времени периодом отбора и недостаточным количеством проанализированных проб воды. Расширение перечня обследуемых водоемов на различных территориях может стать направлением дальнейших исследований по оценке содержания антибиотиков в водных средах.
Заключение. Разработанная методика может быть использована в гигиенических исследованиях содержания остаточных количеств антибиотиков в воде для оценки качества источников питьевого и культурно-бытового водоснабжения.
1. Распоряжение Правительства РФ «О Стратегии предупреждения распространения антимикробной резистентности в РФ на период до 2030 г.». Утверждено распоряжением Правительства Российской Федерации от 25 сентября 2017 г. № 2045-р https://www. garant.ru/products/ipo/prime/doc/71677266
2. СанПиН 1.2.3685–21 «Гигиенические нормативы и требования к обеспечению безопасности и (или) безвредности для человека факторов среды обитания». Утверждены постановлением Главного государственного санитарного врача Российской Федерации от 28 января 2021 года № 2.
3. Пругло Г.Ф., Федорова О.В., Смит Р.А. Хроматографические методы анализа: учебное пособие. СПб.: ВШТЭ СПбГУПТД, 2017. 85 с.
4. Токсикологическая химия: учебник / Т. Байзолданов. – Алматы: Эверо, 2021. 240 с.
5. EPA Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS. 2007.
1. Антропова Н.С., Ушакова О.В., Водянова М.А., Савостикова О.Н. Риск распространения антибиотикорезистентности через объекты окружающей среды и продукты питания (обзорная статья) // Российский журнал восстановительной медицины. 2020. № 4. С. 36–51. EDN EQYXXJ.
2. Mutua F, Sharma G, Grace D, Bandyopadhyay S, Shome B, Lindahl J. A review of animal health and drug use practices in India, and their possible link to antimicrobial resistance. Antimicrob Resist Infect Control. 2020;9(1):103. doi: 10.1186/s13756-020-00760-3
3. Li S, Shi W, You M, et al. Antibiotics in water and sediments of Danjiangkou Reservoir, China: Spatiotemporal distribution and indicator screening. Environ Pollut. 2019;246:435–442. doi: 10.1016/j.envpol.2018.12.038
4. Mijangos L, Ziarrusta H, Ros O, et al. Occurrence of emerging pollutants in estuaries of the Basque Country: analysis of sources and distribution, and assessment of the environmental risk. Water Res. 2018;147:152-163. doi: 10.1016/j.watres.2018.09.033
5. Fernandes JP, Almeida CMR, Salgado MA, Carvalho MF, Mucha AP. Pharmaceutical compounds in aquatic environments – Occurrence, fate and bioremediation prospective. Toxics. 2021;9(10):257. doi: 10.3390/toxics9100257
6. Марченко Б.И., Назарянц А.А., Нестерова О.А. Фармацевтические средства и их дериваты в водных объектах как фактор эколого-гигиенического риска. Анализ риска здоровью – 2023. Совместно с международной встречей по окружающей среде и здоровью RISE-2023: материалы XIII Всероссийской научно-практической конференции с международным участием / под ред. проф. А.Ю. Поповой, акад. РАН Н.В. Зайцевой. Пермь: Изд-во Перм. нац. исслед. политехн. ун-та, 2023. Т. 1. С. 60–68.
7. Bengtsson-Palme J, Kristiansson E, Larsson DG. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol Rev. 2018;42(1):fux053. doi: 10.1093/femsre/fux053
8. Manyi-Loh CE, Okoh AI, Lues R. Occurrence and multidrug resistance in strains of Listeria monocytogenes recovered from the anaerobic co-digestion sludge contained in a single stage steel biodigester: Implications for antimicrobial stewardship. Microorganisms. 2023;11(3):725. doi: 10.3390/microorganisms11030725
9. Maghsodian Z, Sanati AM, Mashifana T, et al. Occurrence and distribution of antibiotics in the water, sediment, and biota of freshwater and marine environments: A review. Antibiotics (Basel). 2022;11(11):1461. doi: 10.3390/antibiotics11111461
10. Huang F, An Z, Moran MJ, Liu F. Recognition of typical antibiotic residues in environmental media related to groundwater in China (2009–2019). J Hazard Mater. 2020;399:122813. doi: 10.1016/j.jhazmat.2020.122813
11. Bianco K, de Farias BO, Gonçalves-Brito AS, et al. Mobile resistome of microbial communities and antimicrobial residues from drinking water supply systems in Rio de Janeiro, Brazil. Sci Rep. 2022;12(1):19050. doi: 10.1038/s41598-022-21040-7
12. Лаврухина О.И., Амелин В.Г., Киш Л.К., Третьяков А.В., Пеньков Т.Д. Определение остаточных количеств антибиотиков в объектах окружающей среды и пищевых продуктах // Журнал аналитической химии. 2022. T. 77. № 11. С. 969–1015.
13. Wang J, Ye K-X, Tian Y, et al. Simultaneous determination of 22 antibiotics in environmental water samples by solid phase extraction-high performance liquid chromatography-tandem mass spectrometry. Se Pu. 2023;41(3):241–249. (In Chinese.) doi: 10.3724/SP.J.1123.2022.06004
14. Qi M, He P, Hu H, et al. An automated solid-phase extraction–UPLC–MS/MS method for simultaneous determination of sulfonamide antimicrobials in environmental water. Molecules. 2023;28(12):4694. doi: 10.3390/molecules28124694
15. Zeng Y, Chang F, Liu Q, Duan L, Li D, Zhang H. Recent advances and perspectives on the sources and detection of antibiotics in aquatic environments. J Anal Methods Chem. 2022;2022:5091181. doi: 10.1155/2022/5091181
16. Chen Y, Xie Q, Wan J, Yang S, Wang Y, Fan H. Occurrence and risk assessment of antibiotics in multifunctional reservoirs in Dongguan, China. Environ Sci Pollut Res Int. 2020;27(12):13565–13574. doi: 10.1007/s11356-019-07436-5
17. Bailón-Pérez MI, García-Campaña A, Cruces-Blanco C, del Olmo Iruela M. Trace determination of β-lactam antibiotics in environmental aqueous samples using off-line and on-line preconcentration in capillary electrophoresis. J Chromatogr A. 2008;1185(2):273–280. doi: 10.1016/j.chroma.2007.12.088
18. Goessens T, Huysman S, De Troyer N, et al. Multi-class analysis of 46 antimicrobial drug residues in pond water using UHPLC-Orbitrap-HRMS and application to freshwater ponds in Flanders, Belgium. Talanta. 2020;220:121326. doi: 10.1016/j.talanta.2020.121326
19. Wang J. Analysis of macrolide antibiotics, using liquid chromatography–mass spectrometry, in food, biological and environmental matrices. Mass Spectrom Rev. 2009;28(1):50–92. doi: 10.1002/mas.20189
20. Hu P, Ma X, Bo T. Determination confirmation quantification of trace ß-lactam antibiotics in milk by LC/MS/ MS. Application, Agilent Technologies. 2009. https://www.manuallib.com/file/2648937
21. Chitescu CL, Kaklamanos G, Nicolau AI, Stolker AA. High sensitive multiresidue analysis of pharmaceuticals and antifungals in surface water using U-HPLC-Q-Exactive Orbitrap HRMS. Application to the Danube river basin on the Romanian territory. Sci Total Environ. 2015;532:501–511. doi: 10.1016/j.scitotenv.2015.06.010
22. Böger B, Surek M, Vilhena RO, et al. Occurrence of antibiotics and antibiotic resistant bacteria in subtropical urban rivers in Brazil. J Hazard Mater. 2021;402:123448. doi: 10.1016/j.jhazmat.2020.123448
23. Ye Z, Weinberg HS, Meyer M. Trace analysis of trimethoprim and sulfonamide, macrolide, quinolone, and tetracycline antibiotics in chlorinated drinking water using liquid chromatography electrospray tandem mass spectrometry. Anal Chem. 2007;79(3):1135–1144. doi: 10.1021/ac060972a
24. Chen Y, Xie Q, Wan J, Yang S, Wang Y, Fan H. Occurrence and risk assessment of antibiotics in multifunctional reservoirs in Dongguan, China. Environ Sci Pollut Res Int. 2020;27(12):13565–13574. doi: 10.1007/s11356-019-07436-5
25. Bianco K, de Farias BO, Gonçalves-Brito A, et al. Mobile resistome of microbial communities and antimicrobial residues from drinking water supply systems in Rio de Janeiro, Brazil. Sci Rep. 2022;12(1):19050. doi: 10.1038/s41598-022-21040-7
26. Azzi M, Ravier S, Elkak A, Coulomb B, Boudenne J-L. Fast UHPLC-MS/MS for the simultaneous determination of azithromycin, erythromycin, fluoxetine and sotalol in surface water samples. Appl Sci. 2021;11(18):8316. doi: 10.3390/app11188316
Нурисламова Татьяна Валентиновна – д.б.н., доцент, заведующая отделом химико-аналитических методов исследования.
ул. Монастырская, д. 82, Пермь, 614045
Карнажицкая Татьяна Дмитриевна – к.б.н., заведующая лабораторией методов жидкостной хроматографии.
ул. Монастырская, д. 82, Пермь, 614045
Старчикова Мария Олеговна – младший научный сотрудник лаборатории методов жидкостной хроматографии.
ул. Монастырская, д. 82, Пермь, 614045
Терентьев Геннадий Ильич – заведующий отделением физико-химических методов исследования ФБУЗ «Центр гигиены и эпидемиологии в Пермском крае».
ул. Монастырская, д. 82, Пермь, 614045; ул. Куйбышева, д. 50, Пермь, 614016
Поспелова Анна Анатольевна – к.фарм.н., химик-эксперт.
ул. Куйбышева, д. 50, Пермь, 614016
Нурисламова Т.В., Карнажицкая Т.Д., Старчикова М.О., Терентьев Г.И., Поспелова А.А. Методические подходы к определению антибиотиков в воде на уровне гигиенических нормативов методом высокоэффективной жидкостной хроматографии / масс-спектрометрии. Здоровье населения и среда обитания – ЗНиСО. 2024;32(2):32-41. https://doi.org/2219-5238/2024-32-2-32-41
Nurislamova T.V., Karnazhitskaya T.D., Starchikova M.O., Terentyev G.I., Pospelova A.A. Methodological Approaches to Determination of Antibiotics in Water at the Level of Hygienic Standards Using High-Performance Liquid Chromatography–Mass Spectrometry. Public Health and Life Environment – PH&LE. 2024;32(2):32-41. (In Russ.) https://doi.org/2219-5238/2024-32-2-32-41
ЖУРНАЛ В СОЦСЕТЯХ
![]()
![]()
.png)
117105, Москва, Варшавское шоссе, 19А
ФБУЗ «Федеральный центр гигиены и эпидемиологии» Роспотребнадзора
Редакция «ЗНиСО»
E-mail: zniso@fcgie.ru






























