




Содержание
Перейти к:
https://doi.org/10.35627/2219-5238/2022-30-9-35-42
Перейти к:
Введение. На развитых металлургических предприятиях зачастую наблюдается превышение предельно допустимых концентраций тяжелых металлов, оказывающих пагубное влияние на здоровье рабочих. Тяжелые металлы вызывают окислительный стресс, являющийся одним из ключевых факторов развития атеросклеротического поражения сосудов. Атеросклероз приводит к сердечным приступам и инсультам, которые являются причиной гибели людей в 85 % случаев смерти от сердечно-сосудистых заболеваний.
Цель: Изучение молекулярных механизмов атеросклероза сосудов и влияния тяжелых металлов на протекание заболевания.
Методы. Использованы информационно-аналитические методы на основе обобщения и анализа современных научных исследований, опубликованных в реферативных базах данных NLM, Scopus, CyberLeninka, Google Scholar, eLibrary, а также в информационных порталах по состоянию на январь 2022 г. Отбор статей осуществлялся по принципу наличия в них сведений о патогенезе и влиянии тяжелых металлов на протекание атеросклероза. Было проанализировано более 400 статей, в результате их них отобрано 66 полнотекстовых материалов.
Результаты. Показана связь между молекулярными механизмами атеросклероза и тяжелыми металлами, сопоставлены основные этапы развития заболевания и метаболомный профиль крови при патологии.
Заключение. Представленный обзор литературы выявил проблемы в нормативной базе и практическом осуществлении гигиенической оценки влияния тяжелых металлов на протекание атеросклеротического поражения сосудов, одной из которых является недостаточная оценка вклада тяжелых металлов в протекание заболевания. На данный момент удалось обнаружить влияние тяжелых металлов лишь на изолированные процессы патогенеза атеросклероза: изменение проницаемости и разрушение сосудистых мембран, повышение окислительного стресса, воспаление, пролиферацию гладкомышечных клеток, изменение реологических свойств крови, повышенный риск тромбообразования. Найденные закономерности в изменении концентрации некоторых метаболитов крови и потенциальное пагубное влияние тяжелых металлов на поражение сосудов позволят в будущем разработать новые способы выявления атеросклероза и включить рабочих промышленных предприятий в группу риска для ранней диагностики заболевания.
Унесихина М.С., Чемезов А.И., Сутункова М.П. Метаболомное профилирование при атеросклеротическом поражении сосудов и влияние тяжелых металлов на протекание заболевания (обзор литературы). Здоровье населения и среда обитания – ЗНиСО. 2022;(9):35-42. https://doi.org/10.35627/2219-5238/2022-30-9-35-42
Unesikhina M.S., Chemezov A.I., Sutunkova M.P. Metabolomic Profiling in Atherosclerotic Lesions and the Effect of Heavy Metals on the Course of Disease: A Literature Review. Public Health and Life Environment – PH&LE. 2022;(9):35-42. (In Russ.) https://doi.org/10.35627/2219-5238/2022-30-9-35-42
Введение. По данным Всемирной организации здравоохранения (ВОЗ), ССЗ являются ведущей причиной гибели людей во всем мире. По оценкам ВОЗ в 2019 году от ССЗ умерло 17,9 миллиона человек, что составляет 32 % всех смертей в мире, из них 85 % вызваны сердечным приступом и инсультом [1]. Сердечные приступы и инсульты в основном являются следствием закупорки сосудов, что препятствует кровоснабжению жизненно необходимых органов. Наиболее распространенной причиной этого является накопление жировых отложений на внутренних стенках кровеносных сосудов, так называемый атеросклероз.
Несмотря на то что в мире и, в частности, в странах с развитой металлургической промышленностью, особенно в РФ, наблюдается положительная тенденция к снижению числа смертей от ССЗ (см. рисунок), число сохраненных жизней благодаря современной науке необходимо наращивать более стремительными темпами.
Так как в Свердловской области широко представлена такая отрасль промышленности, как металлургические предприятия, это приводит к загрязнению окружающей среды: в частности, повышенное содержание тяжелых металлов оказывает заметное пагубное влияние на рабочих таких производств. Основными контаминантами среди тяжелых металлов являются свинец, кадмий, железо, никель, медь, мышьяк и ртуть, которые чаще других элементов превышают нормативные показатели в Свердловской области [3]. На текущий момент уже были проведены исследования, демонстрирующие прямую корреляцию между концентрациями свинца и кадмия в крови людей и повышенной смертностью, особенно от сердечно-сосудистых заболеваний (ССЗ) [4][5]. Также среди всех регионов РФ была показана прямая зависимость между занятостью мужчин во вредных и опасных условиях труда и показателями смертности от ССЗ [6].
Атеросклеротическое поражение диагностируют при появлении клинических симптомов, после липидного профилирования крови и применения инструментальных методов исследования (компьютерная томография, измерение толщины интимы сонной артерии, внутрисосудистое ультразвуковое исследование), когда опасность возникновения осложнений может быть уже высока, поэтому необходим метод ранней и простой диагностики до появления характерных признаков поражения сосудов у людей, входящих в группу риска и склонных к дислипидемиям.
Целью данной обзорной статьи является изучение молекулярных механизмов атеросклероза сосудов и вклада экспозиции к тяжелым металлам в протекание заболевания. Анализ данной информации позволит выявить закономерности в изменении метаболомного профиля крови для разработки методики ранней диагностики атеросклеротического поражения.
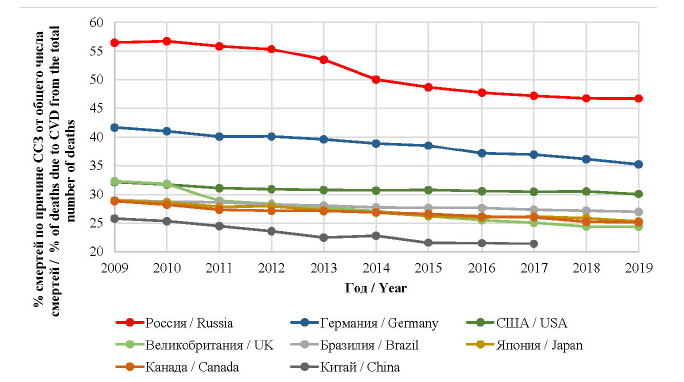
Рисунок. Доля смертей по причине сердечно-сосудистых заболеваний от общего числа смертей
в странах с развитой металлургической промышленностью в 2009–2019 гг. [2]
Figure. The proportion of deaths from cardiovascular diseases of the total deaths registered in the countries with a developed
metallurgical industry, 2009–2019 [2]
Материалы и методы. Использованы информационно-аналитические методы на основе обобщения и анализа современных научных исследований, опубликованных в реферативных базах данных NLM, Scopus, CyberLeninka, Google Scholar, eLibrary, а также информационных порталах по состоянию на январь 2022 г. Отбор статей осуществлялся по принципу наличия в них сведений о влиянии тяжелых металлов на протекание атеросклероза и метаболомного профилирования людей и животных с атеросклерозом. Было проанализировано более 400 оригинальных статей, и в результате их них отобрано 66 полнотекстовых материалов.
Результаты и обсуждение. В статье показана связь между молекулярными механизмами атеросклеротического поражения сосудов и влиянием тяжелых металлов. В частности, были сопоставлены основные этапы развития заболевания и результаты метаболомного профилирования крови как людей, так и животных.
Диагностические возможности
Многие изменения состава крови, наблюдаемые при атеросклеротических поражениях, касаются метаболомного профиля крови – совокупности метаболитов, т. е. низкомолекулярных веществ с массой до 1500 Да, являющихся конечными и промежуточными продуктами метаболизма.
Главными аналитическими технологиями в области метаболомики является масс-спектрометрия (МС) в сочетании с методами разделения, самой распространенной и универсальной из которых является высокоэффективная жидкостная хроматография (ВЭЖХ), обладающая большой специфичностью, высокой скоростью анализа и возможностью точного количественного определения искомых соединений. Благодаря таким мощным аналитическим методам простой анализ крови может помочь в быстрой диагностике атеросклероза.
Атеросклероз
Атеросклероз – хроническое воспалительное заболевание средних и крупных артерий, сопровождающееся инфильтрацией очага поражения липидами и иммунными клетками, разрастанием соединительной ткани с образованием фиброзных колпачков и последующим сужением просвета сосудов, что приводит к общему нарушению кровообращения и риску разрыва бляшки с формированием тромба.
Возраст, наследственность, дислипидемия, артериальная гипертензия, повышенное употребление пищи, обогащенной насыщенными жирными кислотами и холестерином, ожирение, курение, сахарный диабет, малоактивный образ жизни, гипергомоцистеинемия – факторы, способствующие инициации и прогрессированию атеросклероза [7]. Липопротеины (ЛП), являющиеся биохимическим комплексом, транспортирующим триглицериды (ТГ), фосфолипиды и холестерин (ХС), показывают прямую корреляцию между концентрацией их в крови и атеросклерозом. Доказано, что высокие уровни липопротеинов низкой плотности (ЛПНП), липопротеинов очень низкой плотности (ЛПОНП), ТГ, ХС и низкий уровень липопротеинов высокой плотности (ЛПВП) являются ведущими предикторами появления патологии.
Липидная инфильтрация стенки артерий
Атеросклероз начинается как воспалительная реакция на задержку ЛПНП в интиме сосудов, что переходит в последствии в порочный круг – «реакция на удержание».
В обычных условиях ламинарный кровоток способствует образованию эндотелиального релаксирующего фактора (NO), который оказывает сосудорасширяющее действие и обладает противовоспалительной активностью, поэтому патологическим изменениям наиболее подвержены искривленные участки артерий, испытывающие гемодинамический стресс растяжения в местах нарушения ламинарного потока крови. Заметное влияние на снижение уровня NO было замечено у свинца [8], кадмия [9] и железа [10][11], что в условиях увеличения содержания АФК нарушает гомеостатические механизмы сосудов, способствует агрегации тромбоцитов, сужению сосудов и воспалительному диапедезу.
Это приводит к травмированию сосуда с последующей активацией эндотелия (усиливается локальный синтез протеогликанов с высоким сродством к ЛП). Эндотелиоциты начинают экспрессировать на поверхности мембраны молекулы адгезии, интегрины и селектины, приводящие к утолщению интимы [12], и секретировать интерлейкины, интерфероны и другие цитокины [13], запуская воспалительную реакцию.
Таким образом, ЛПНП, имеющие диаметр до 70 нм, легко проникающие в эндотелий посредством трансцитоза и диффундирующие через ткань в обычных условиях, начинают прилипать к стенке сосуда за счет связывания положительно заряженного остатка белка Апо-В100 ЛПНП с отрицательно заряженными цепями глюкозаминогликанов в протеогликанах (компоненты соединительной ткани стенки артерий). Такие необратимые изменения конформации белка делают частицы ЛПНП более восприимчивыми к модификациям, что затрудняет их отток [14].
Удерживание ЛПНП в эндотелии сосудов также усиливают сфингомиелинфосфодиэстераза (SMAse) и липопротеинлипаза (положительно заряженный фермент, который обычно связан с отрицательно заряженными протеогликанами в просвете сосуда, где захватывает циркулирующие липопротеины (ЛП)), секретируемые макрофагами. Комплексы ЛП-протеогликан, в сравнении с несвязанными ЛПНП, более склонны к окислению и агрегации. В свою очередь, модифицированные таким образом ЛПНП усиливают выработку протеогликанов с высоким сродством к ЛП [15].
Накопленные ЛПНП взаимодействуют с активными формами кислорода (АФК) и ферментами миелопероксидазой и липоксигеназой, выделяемыми воспалительными клетками [16]. Окисленные ЛПНП (оЛПНП) становятся более подверженными изменению конформации белка АпоВ-100, в результате чего у них повышается сродство к рецепторам – поглотителям макрофагов. Значительный вклад в окисление липидов вносят тяжелые металлы благодаря своей способности образовывать свободные радикалы и индуцировать перекисное окисление липидов (ПОЛ). В результате окисления фосфолипидов мембран изменяется их целостность, проницаемость и текучесть [17].
В оЛПНП основными фосфолипидами являются лизофосфатидилхолины (LPC), тогда как в немодифицированных ЛПНП LPC составляют лишь 1–5 % от общего числа фосфатидилхолинов (PC) [18]. Повышенные концентрации LPC в крови при атеросклеротических поражениях наблюдаются во многих исследованиях [19–23].
Избыточные количества LPC образуются за счет увеличения экспрессии секреторной фосфолипазы А2 группы 2 (sPLA2-II), которая расщепляет PC в оЛПНП до LPC и жирных кислот. sPLA2-II экспрессируется в клетках и тканях, участвующих в иммунных и воспалительных ответах: макрофаги, тучные клетки, фибробласты, ткани печени, селезенки, тимуса, костного мозга. Избыточная активность sPLA2-II индуцируется в условиях повышенного содержания провоспалительных цитокинов, гипоксии и других факторов воспаления, которые наблюдаются при атеросклеротическом поражении сосудов. Также значительное влияние на повышение активности sPLA2-II дозозависимым образом оказывает ртуть, способствуя таким образом развитию атеросклероза [24]. sPLA2-II усиливает окисление ЛПНП, индуцированное 15-липоксигеназой, и также благоприятствует слиянию ЛПНП с протеогликанами за счет их окислительной модификации [25].
Воспалительный ответ
Накопленные в атеросклеротическом поражении LPC индуцируют экспрессию хемокинов и молекул адгезии лейкоцитов ЭК сосудов и ГМК при участии моноцитарного хемокинового белка 1 (MCP-1). MCP-1 ответствен за хемотаксис моноцитов в стенку артерий и их дифференциацию в макрофаги за счет активации рецепторов CCR2 и образования эндотелиальных молекул адгезии лейкоцитов (белок адгезии сосудистых клеток (VCAM-1) и молекула клеточной адгезии (ICAM-1)) [26]. Подобное воздействие на организм оказывает железо: в исследованиях были замечены повышенные концентрации проатеросклеротических хемокинов/цитокинов (MCP-1, IL-6 и TNF-α) и молекул адгезии (ICAM-1, VCAM-1), т. к. железо имеет свойство накапливаться в медиальном слое ЭК и ГМК сосудов [10]. Уровень VCAM-1 также заметно повышался при воздействии наночастиц никеля на организм [27].
LPC опосредуют ремоделирование сосудистых клеток за счет стимулирования продукции провоспалительных цитокинов (IL-1, IL-6, IL-8) моноцитами, индуцируют окислительный стресс с образованием АФК и увеличивают экспрессию рецепторов-поглотителей, способствуя захвату оЛПНП макрофагами и образованию пенистых клеток [28][29]. Было продемонстрировано, что не все LPC являются атерогенными: так, провоспалительными и атерогенными свойствами облают LPC с длинной и насыщенной ацильной цепью не менее 16 углеродов [28].
Во время дифференцировки моноцитов в макрофаги экспрессируются рецепторы-мусорщики (CD36, SR-AI/II и SR-BI) и лектин-подобный окислительный рецептор ЛПНП (LOX-1), которые отвечают за распознавание оЛПНП макрофагами [29]. Сами же оЛПНП, в отличие от немодифицированных ЛПНП, более активно поглощаются макрофагами за счет изменения конформации своей белковой части и их более высокого сродства к рецепторам – поглотителям макрофагов [12], что также сопровождается неспособностью рецепторов ЛПНП распознавать и транспортировать обратно в печень такие оЛПНП.
Участие аминокислот в атеросклеротическом поражении
Вклад гомоцистеина в развитие атеросклероза также немаловажен. Повышенный уровень гомоцистеина, который может быть обусловлен генетическими нарушениями ферментов, участвующими в метаболизме гомоцистеина, дефицитом коферментов и избыточным поступлением метионина из пищевого белка, значительно повышает риск атеросклеротического поражения сосудов [19][30]. Установлено, что гомоцистеин участвует в изменении окислительно-восстановительной сигнализации клеток, индуцируя НАДФН-оксидазу и активность эндотелиальной синтазы оксида азота (eNOS), что приводит к окислительному повреждению клеток и эндотелиальной дисфункции соответственно [31]. eNOS из-за высокого окислительного стресса экспрессирует пероксинитрит, вызывающий ПОЛ, что также усугубляет нарушения редокс-статуса организма. Прооксидантное состояние способствует стрессу эндоплазматического ретикулума (ER-стрессу) и активации медиатора воспаления NF-kB, который ответственен за активацию ЭК и инициирование экспрессии МСР-1[32].
Дополнительно можно сделать предположение, что повышение уровня метионина и бетаина в исследованиях [33][34] происходит в результате компенсаторного механизма для снижения концентрации гомоцистеина. Помимо же этого, бетаин является продуктом разложения PC, чьи повышенные концентрации в крови замечены при атеросклерозе [35].
Метионин и гомоцистеин являются серосодержащими аминокислотами, легко подвергающимися окислению, тем более в присутствии тяжелых металлов, являющихся «тиоловыми ядами», которые блокируют сульфгидрильные группы белков, ферментов и аминокислот, участвующих в антиоксидантной защите организма.
Несмотря на описанное участие многих метаболитов в прогрессировании атеросклеротических поражений, заметное снижение уровня триптофана [19][33][36][37] является в некотором проявлении антиатерогенным механизмом, обусловленным избыточной экспрессией индоламин-2,3-диоксигеназы (IDO) с последующим катаболизмом триптофана, т. е. происходит активация кинуренинового пути [38]. Повышенная экспрессия IDO связана с хроническим воспалением низкой степени тяжести, характерным для атеросклероза. Так, было показано, что белок IDO экспрессируется антигенпрезентирующими клетками, включая макрофаги и дендритные клетки, сильно индуцируется провоспалительными цитокинами в атеросклеротических бляшках и активируется в клетках, ассоциированных с атеромой [39]. Повышенное соотношение кинуренин/триптофан было связано с утолщением сонных артерий и повышенными концентрациями ЛПНП, ТГ и ХС [40][41]. Согласно теории истощения триптофана, активация IDO в антигенпрезентирующих клетках лишает Т-клетки этой незаменимой аминокислоты, тем самым подавляя их активность и пролиферацию, что снижает воспаление сосудов и прогрессирование атеросклероза [39].
Было продемонстрировано влияние 3-гидроксиантраниловой кислоты, продукта катаболизма триптофана, на снижение уровня ЛПНП, ТГ, ХС и повышение уровня ЛПВП и замечены ее антиоксидантные и противовоспалительные свойства (снижение уровня VCAM-1, MCP-1). Но некоторые метаболиты кинуренинового пути проявляют и атерогенные свойства, например кинуренин, 3-гидроксикинуренин и хинолиновая кислота способствуют образованию супероксидных радикалов [38].
Нарушение энергетического обмена
Помимо прочего, было показано, что присутствие локализованной гипоксии и АФК в поврежденных сосудах являются одним из стимулов дифференцировки моноцитов в макрофаги, в частности через активацию провоспалительных генов [42], что также облегчает превращение макрофагов в пенистые клетки. Ключевым медиатором эффектов, опосредуемых гипоксией, является транскрипционный фактор, индуцируемый гипоксией 1-альфа (HIF-1α). При низких концентрациях кислорода происходит увеличение времени жизни белка HIF-1α, который способствует пролиферации ГМК и рекрутированию моноцитов, других лейкоцитов и Т-лимфоцитов в бляшку, снижает миграционную активность макрофагов и удерживает их в интиме [7][13]. Таким образом, популяция макрофагов и пенистых клеток постоянно растет.
Так как сосудистые нарушения характеризуются гипоксией, макрофаги переходят на гликолитический тип метаболизма, хоть он и является менее энергоэффективным [42][43]. В условиях же сниженного содержания АТФ накапливается АМФ, стимулирующая β-окисление [7]. Параллельно из-за недостатка кислорода и повышенной потребности в энергии происходит нарушение самого β-окисления, процесса, протекающего в аэробных условиях. В результате митохондрии перегружаются, что ведет к накоплению ацилкарнитинов, свободных жирных кислот, ацетил-КоА и снижению концентрации L-ацетилкарнитина [44]. Было показано, что ацилкарнитины, накапливающиеся при атеросклеротических поражениях [20][22][45], индуцируют экспрессию провоспалительных факторов дозозависимым образом, включая IL-1β, IL-6, фактор некроза опухоли α (TNF-α), MCP-1 и ядерный фактор «каппа би» (NF-kB) [46].
Немаловажный вклад в истощение энергетических запасов клеток вносит мышьяк за счет своей способности замещать фосфат, встраиваясь в фосфодиэфирную связь АТФ, что приводит к расщеплению цепи окислительного фосфорилирования и последующему истощению запасов АТФ [17].
В результате нарушения β-окисления снижается образование ацетил-КоА, значительная доля которого поступает в цикл трикарбоновых кислот (ЦТК). Для восполнения дефицита активируются анаплеротические пути поддержания ЦТК, субстратами которых служат аминокислоты (в т. ч. серин, триптофан), что также может объяснять их пониженные концентрации при атеросклеротических поражениях [7][20][36].
Участие липидов в атеросклеротическом поражении
Клеточный ответ на насыщенные жирные кислоты, чьи повышенные концентрации наблюдаются при атеросклерозе [33][34][47][48], опосредуется врожденной иммунной системой за счет воздействия на толл-подобный рецептор 4 (TLR4) с последующей экспрессией TNF-α и MCP-1 [49]. Было показано, что жирные кислоты являются источниками АФК за счет активации НАДФН-оксидазы и нарушают продукцию и выработку NO, отвечающего за вазодилатацию сосудов [50]. К тому же они активируют криопирин (NLRP3), который усиливает генерацию АФК в макрофагах и индукцию синтеза провоспалительных цитокинов (IL-1β и IL-18) [51].
Атеросклеротическая бляшка характеризуется разнообразием липидов, высвобождаемых из ЛПНП. Так, помимо LPC и жирных кислот, замечены повышенные концентрации церамидов [35][52][53], фитосфингозина и сфинганина [21][47], что, вероятно, является следствием действия SMAse, которая расщепляет сфингомиелины на поверхности ЛП. Было показано, что активность SMAse стимулирует TNF-α [52]. В неагрегированных и неокисленных ЛПНП церамидов обнаружено не было [54].
Церамиды активируют НАДФН-оксидазу с последующим увеличением содержания супероксидных анионов [52], индуцируют ER-стресс и блокируют β-окисление митохондрий. К тому же они участвуют в трансцитозе оЛПНП через ЭК, способствуя удержанию липидов в сосудистой стенке и вызывая конформационные изменения в АpoB-100 [55].
Пролиферация и миграция ГМК
Параллельно со всем происходящим за счет выделения цитокинов и факторов роста вышеперечисленными участниками атеросклеротических поражений, вызывающих рост клеток и образование белков внеклеточного матрикса (коллаген, эластин, протеогликан), происходит миграция ГМК в интиму. Такие мигрировавшие ГМК и синтезированный внеклеточный матрикс образуют фиброзный колпачок, который состоит из богатых коллагеном волокнистых тканей, ГМК, макрофагов и Т-лимфоцитов. Это объясняется тем, что ГМК в случае повреждения способны переключаться с «нормального сократительного фенотипа» на «провоспалительный-синтетический». Это изменение приводит к снижению экспрессии белков, ответственных за сокращение ГМК, и повышению продукции провоспалительных медиаторов, которые запускают пролиферацию и миграцию ГМК [56].
Вместе с этим после воздействия ртути и железа на организм была замечена повышенная экспрессия фактора роста эндотелия сосудов (VEGF) в ЭК, который участвует в их миграции, пролиферации и созревании [10][57].
Также было отмечено, что слой протеогликанов в интиме, имеющий повышенное содержание коллагена и измененную продольную ориентацию коллагеновых волокон, был в 4 раза толще в областях атеросклеротического поражения и являлся основным фактором стеноза артерий [58].
Некротическое ядро
Многие механизмы развития атеросклероза могут инициировать поступающие в организм тяжелые металлы: их основной механизм влияния – окислительный стресс, являющийся главным патогенетическим фактором поражения сосудов. Так, в исследованиях кадмий [59][9], свинец[59][60], железо [10][11], никель [27], медь [11], мышьяк и ртуть [17] вызывали окислительный стресс с увеличением продуктов ПОЛ (наблюдался высокий уровень малонового диальдегида и оксистеролов), оЛПНП и АФК преимущественно за счет образования высокоактивных свободных радикалов по реакции Фентона и истощения запасов внутриклеточных антиоксидантов и участвующих в их синтезе ферментов, по причине высокого сродства к сульфгидрильным группам.
При запущенных поражениях окислительный стресс в макрофагах, ЭК и ГМК может привести к их апоптозу, который запускается через ER-стресс. ER-стресс представляет собой реакцию клеток на временные или длительные нарушения функций ER, особенно функций, связанных с синтезом белка, регуляцией кальция и внутриклеточным окислительно-восстановительным потенциалом. При продолжительном состоянии ER-стресса включается развернутый белковый ответ (UPR) в качестве защитного механизма от перегрузки [61]. К стрессорам, которые активируют UPR, относятся окислительный стресс, гомоцистеин, гипоксия, оксистеролы, высокие уровни внутриклеточного холестерина и насыщенных жирных кислот [61]. UPR приводит к повышению уровня цитозольного кальция и активации кальций/ кальмодулин-зависимой протеинкиназы типа II (CaMKII). CaMKII запускает апоптоз с помощью экспрессии НАДФН-оксидазы и последующей генерации АФК. Вторым необходимым механизмом для апоптоза служат рецепторы комбинированного распознавания образов (PRRs), которые связывают патогены, чужеродные антигены, эндогенные белки и модифицированные липиды [62]. В результате со временем соседние фагоциты перестают успевать очищать место поражения от апоптотических клеток, что приводит к разрастанию некротического ядра и увеличению площади атероматозного поражения, способствуя тем самым истончению фиброзной оболочки и разрушению бляшки.
Нестабильность атеросклеротической бляшки
Накопление всех этих факторов образует зрелую атеросклеротическую бляшку в интиме сосуда, уменьшая его кровоток. Но прочность бляшки может быть нарушена, когда механическое напряжение в волокнистой оболочке превышает критический уровень, который может выдержать ткань. Среди причин, способствующих разрыву бляшки, можно выделить истончение фиброзной оболочки, разрастающееся некротическое ядро и матриксные металлопротеиназы 2 и 9 (ММП-2 и ММП-9), которые секретируются макрофагами и АФК [29], что приводит к разрушению внеклеточного матрикса.
Стабильность атеросклеротической бляшки может также изменять кальцификация и неоваскуляризация – черты уже глубоко прогрессирующих поражений. Кальцификация интимы связана с перицитоподобными клетками, секретирующими компоненты матрикса, которые впоследствии подвергаются кальцификации (процесс, регулируемый оксистеролами и цитокинами) [63]. Кальцификация сосудов также может происходить на фоне активации матриксной ММП-9 и сочетаться с деградацией эластина, метаболиты которого способны дополнительно активировать клеточно-зависимое отложение кальция [64]. Повышенная потребность в кислороде приводит к васкуляризации, но образование новых сосудов сконцентрировано на границах бляшки, в то время как центральная часть находится в условиях постоянной гипоксии, что объясняет многие наблюдаемые метаболические изменения [65].
Тромбоз, основное осложнение атеросклероза, связан с разрывом бляшки, в результате чего запускается каскад свертывания крови, вызывая образование тромба, который может проникнуть в просвет сосуда, где он будет блокировать кровоток. Процесс тромбообразования также способна стимулировать ртуть, вызывая активацию тромбоцитов, увеличивая активность факторов свертывания крови и усиливая адгезию эритроцитов к ЭК за счет изменения формы эритроцитов под влиянием этого металла [57]. Кроме того, тромб может отрываться, вызывая ишемию органов и тканей [66].
Заключение. Влияние тяжелых металлов на протекание атеросклероза существенно – оно способствует прогрессированию заболевания на каждом этапе: от изменения проницаемости и разрушения сосудистых мембран до повышенного содержания АФК и ПОЛ, приводящего к окислительному стрессу, воспалительному ответу, пролиферации ГМК и изменению реологических свойств крови с повышением риска тромбообразования. Замкнутый круг удержания липидов, рекрутирования новых моноцитов, утолщения интимы, гипоксии, окислительного стресса и апоптоза ведет к разрастанию атеросклеротической бляшки, риску ее разрыва и образованию тромба.
Описанные механизмы развития атеросклеротического поражения приводят к характерному изменению метаболомного профиля крови, который охватывает многие процессы обмена веществ. Именно одновременное изменение концентрации таких, как, казалось бы на первый взгляд, неспецифических метаболитов для атеросклероза, как аминокислоты (гомоцистеин, метионин, серин, бетаин, триптофан), жирные кислоты, ацетилкарнитины, глицерофосфолипиды (лизофосфатидилхолины) и сфинголипиды (церамиды, сфинганин, фитосфингозин), может еще на ранних стадиях заболевания свидетельствовать о его начале и служить дополнительным методом диагностики, подтверждающим атеросклеротическое поражение сосудов.
Метаболомный скрининг комплекса вышеобозначенных соединений с помощью современного и высокоточного оборудования на ранних стадиях заболевания может предсказать его наличие, помочь в отслеживании эффективности лечения, а также предоставить новые рычаги воздействия для предотвращения осложнений, которые влечет за собой зрелая атеросклеротическая бляшка. Такой мониторинг метаболомного профиля крови будет крайне важен в том числе для рабочих промышленных предприятий, подвергающихся хронической интоксикации тяжелыми металлами, что повышает степень кардиориска независимо от других отягчающих обстоятельств их образа жизни и здоровья.
1. WHO. Cardiovascular diseases (CVDs). 11 June 2021. Accessed June 21, 2022. https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
2. WHO. Who mortality database; 2022. Accessed June 21, 2022. https://platform.who.int/mortality/themes/theme-details/topics/topic-details/MDB/cardiovascular-diseases
3. Зубков В.А., Тришевская А.В., Малкова Е.А., Михеева Е.В. Содержание тяжелых металлов в почвах промышленных городов Свердловской области. Colloquium-journal. 2019. № 17-7 (41). С. 16–23.
4. Weisskopf MG, Jain N, Nie H, et al. A prospective study of bone lead concentration and death from all causes, cardiovascular diseases, and cancer in the Department of Veterans Affairs Normative Aging Study. Circulation. 2009;120(12):1056-1064. doi: 10.1161/CIRCULATIONAHA.108.827121
5. Tellez-Plaza M, Guallar E, Howard BV, et al. Cadmium exposure and incident cardiovascular disease. Epidemiology. 2013;24(3):421-429. doi: 10.1097/EDE.0b013e31828b0631
6. Бухтияров И.В., Измеров Н.Ф., Тихонов Г.И. и др. Условия труда как фактор повышения смертности в трудоспособном возрасте. Медицина труда и промышленная экология. 2017. № 8. С. 43–49.
7. Кольман Я., Рём К.-Г. Наглядная биохимия. М.: Лаборатория знаний, 2019. 509 с.
8. Dursun N, Arifoglu C, Süer C, Keskinol L. Blood pressure relationship to nitric oxide, lipid peroxidation, renal function, and renal blood flow in rats exposed to low lead levels. Biol Trace Elem Res. 2005;104(2):141-149. doi: 10.1385/BTER:104:2:141
9. Oliveira TF, Batista PR, Leal MA, et al. Chronic cadmium exposure accelerates the development of atherosclerosis and induces vascular dysfunction in the aorta of ApoE-/- mice. Biol Trace Elem Res. 2019;187(1):163-171. doi: 10.1007/s12011-018-1359-1
10. Vinchi F, Porto G, Simmelbauer A, et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J. 2020;41(28):2681-2695. doi: 10.1093/eurheartj/ehz112
11. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2-3):65-87. doi: 10.1016/j.tox.2011.03.001
12. Zmysłowski A, Szterk A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017;16(1):188. doi: 10.1186/s12944-017-0579-2
13. Bogomolova AM, Nikitin AA, Orlov SV, et al. Hypoxia as a factor involved in the regulation of the apoA-1, ABCA1, and complement C3 gene expression in human macrophages. Biochemistry (Mosc). 2019;84(5):529-539. doi: 10.1134/S0006297919050079
14. Hultgardh-Nilsson A, Borén J, Chakravarti S. The small leucine-rich repeat proteoglycans in tissue repair and atherosclerosis. J Intern Med. 2015;278(5):447-461. doi: 10.1111/joim.12400
15. Devlin CM, Leventhal AR, Kuriakose G, Schuchman EH, Williams KJ, Tabas I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler Thromb Vasc Biol. 2008;28(10):1723-1730. doi: 10.1161/ATVBAHA.108.173344
16. Silverstein RL. Inflammation, atherosclerosis, and arterial thrombosis: role of the scavenger receptor CD36. Cleve Clin J Med. 2009;76(Suppl 2):S27-S30. doi: 10.3949/ccjm.76.s2.06
17. Jan AT, Azam M, Siddiqui K, Ali A, Choi I, Haq QM. Heavy metals and human health: Mechanistic insight into toxicity and counter defense system of antioxidants. Int J Mol Sci. 2015;16(12):29592-29630. doi: 10.3390/ijms161226183
18. Chen L, Liang B, Froese DE, et al. Oxidative modification of low density lipoprotein in normal and hyperlipidemic patients: effect of lysophosphatidylcholine composition on vascular relaxation. J Lipid Res. 1997;38(3):546-553.
19. Zhang F, Jia Z, Gao P, et al. Metabonomics study of atherosclerosis rats by ultra fast liquid chromatography coupled with ion trap-time of flight mass spectrometry. Talanta. 2009;79(3):836-844. doi: 10.1016/j.talanta.2009.05.010
20. Tomas L, Edsfeldt A, Mollet IG, et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur Heart J. 2018;39(24):2301-2310. doi: 10.1093/eurheartj/ehy124
21. Liu YT, Peng JB, Jia HM, et al. UPLC-Q/TOF MS standardized Chinese formula Xin-Ke-Shu for the treatment of atherosclerosis in a rabbit model. Phytomedicine. 2014;21(11):1364-1372. doi: 10.1016/j.phymed.2014.05.009
22. Izidoro MA, Cecconi A, Panadero MI, et al. Plasma metabolic signature of atherosclerosis progression and colchicine treatment in rabbits. Sci Rep. 2020;10(1):7072. doi: 10.1038/s41598-020-63306-y
23. Stübiger G, Aldover-Macasaet E, Bicker W, et al. Targeted profiling of atherogenic phospholipids in human plasma and lipoproteins of hyperlipidemic patients using MALDI-QIT-TOF-MS/MS. Atherosclerosis. 2012;224(1):177-186. doi: 10.1016/j.atherosclerosis. 2012.06.010
24. Mazerik JN, Mikkilineni H, Kuppusamy VA, et al. Mercury activates phospholipase a(2) and induces formation of arachidonic acid metabolites in vascular endothelial cells. Toxicol Mech Methods. 2007;17(9):541-557. doi: 10.1080/15376510701380505
25. Kougias P, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Lysophosphatidylcholine and secretory phospholipase A2 in vascular disease: mediators of endothelial dysfunction and atherosclerosis. Med Sci Monit. 2006;12(1):RA5-RA16.
26. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410-1422. doi: 10.1038/nm.2538
27. Kang GS, Gillespie PA, Gunnison A, Moreira AL, Tchou-Wong KM, Chen LC. Long-term inhalation exposure to nickel nanoparticles exacerbated atherosclerosis in a susceptible mouse model. Environ Health Perspect. 2011;119(2):176-181. doi: 10.1289/ehp.1002508
28. Akerele OA, Cheema SK. Fatty acyl composition of lysophosphatidylcholine is important in atherosclerosis. Med Hypotheses. 2015;85(6):754-760. doi: 10.1016/j.mehy.2015.10.013
29. Khatana C, Saini NK, Chakrabarti S, et al. Mechanistic insights into the oxidized low-density lipoprotein-induced atherosclerosis. Oxid Med Cell Longev. 2020;2020:5245308. doi: 10.1155/2020/5245308
30. Ragino YuI, Chernjavski AM, Polonskaya YaV, et al. Oxidation and endothelial dysfunction biomarkers of atherosclerotic plaque instability. Studies of the vascular wall and blood. Bull Exp Biol Med. 2012;153(3):331-335. doi: 10.1007/s10517-012-1708-6
31. Zhou J, Austin RC. Contributions of hyperhomocysteinemia to atherosclerosis: Causal relationship and potential mechanisms. Biofactors. 2009;35(2):120-129. doi: 10.1002/biof.17
32. Refsum H, Smith AD, Ueland PM, et al. Facts and recommendations about total homocysteine determinations: an expert opinion. Clin Chem. 2004;50(1):3-32. doi: 10.1373/clinchem.2003.021634
33. Zha W, A J, Wang G, et al. Metabonomic characterization of early atherosclerosis in hamsters with induced cholesterol. Biomarkers. 2009;14(6):372-380. doi: 10.1080/13547500903026401
34. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57-63. doi: 10.1038/nature09922
35. Jové M, Ayala V, Ramírez-Núñez O, et al. Lipidomic and metabolomic analyses reveal potential plasma biomarkers of early atheromatous plaque formation in hamsters. Cardiovasc Res. 2013;97(4):642-652. doi: 10.1093/cvr/cvs368
36. Teul J, Rupérez FJ, Garcia A, et al. Improving metabolite knowledge in stable atherosclerosis patients by association and correlation of GC-MS and 1H NMR fingerprints. J Proteome Res. 2009;8(12):5580-5589. doi: 10.1021/pr900668v
37. Cason CA, Dolan KT, Sharma G, et al. Plasma microbiome-modulated indole- and phenyl-derived metabolites associate with advanced atherosclerosis and postoperative outcomes. J Vasc Surg. 2018;68(5):1552-1562.e7. doi: 10.1016/j.jvs.2017.09.029
38. Wang Q, Liu D, Song P, Zou MH. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front Biosci (Landmark Ed). 2015;20(7):1116-1143. doi: 10.2741/4363
39. Niinisalo P, Oksala N, Levula M, et al. Activation of indoleamine 2,3-dioxygenase-induced tryptophan degradation in advanced atherosclerotic plaques: Tampere vascular study. Ann Med. 2010;42(1):55-63. doi: 10.3109/07853890903321559
40. Polyzos KA, Ketelhuth DF. The role of the kynurenine pathway of tryptophan metabolism in cardiovascular disease. An emerging field. Hamostaseologie. 2015;35(2):128-136. doi: 10.5482/HAMO-14-10-0052
41. Song P, Ramprasath T, Wang H, Zou MH. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell Mol Life Sci. 2017;74(16):2899-2916. doi: 10.1007/s00018-017-2504-2
42. Strehl C, Fangradt M, Fearon U, Gaber T, Buttgereit F, Veale DJ. Hypoxia: how does the monocyte– macrophage system respond to changes in oxygen availability? J Leukoc Biol. 2014;95(2):233-241. doi: 10.1189/jlb.1212627
43. Будихина А.С., Пащенков М.В. Роль гликолиза в иммунном ответе // Иммунология. 2021. Т. 42. № 1. С. 5–20. doi: 10.33029/0206-4952-2021-42-1-5-20
44. Коткина Т.И., Титов В.Н., Пархимович Р.М. Иные представления о β-окислении жирных кислот в пероксисомах, митохондриях и кетоновые тела. Диабетическая, ацидотическая кома как острый дефицит ацетил-КоА и АТФ // Клиническая лабораторная диагностика. 2014. Т. 59. № 3. С. 14–23.
45. Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J. 2012;163(5):844-850.e1. doi: 10.1016/j.ahj.2012.02.005
46. Rutkowsky JM, Knotts TA, Ono-Moore KD, et al. Acylcarnitines activate proinflammatory signaling pathways. Am J Physiol Endocrinol Metab. 2014;306(12):E1378-E1387. doi: 10.1152/ajpendo.00656.2013
47. Gao X, Ke C, Liu H, et al. Large-scale metabolomic analysis reveals potential biomarkers for early stage coronary atherosclerosis. Sci Rep. 2017;7(1):11817. doi: 10.1038/s41598-017-12254-1
48. Chen X, Liu L, Palacios G, et al. Plasma metabolomics reveals biomarkers of the atherosclerosis. J Sep Sci. 2010;33(17-18):2776-2783. doi: 10.1002/jssc.201000395
49. Blair HC, Sepulveda J, Papachristou DJ. Nature and nurture in atherosclerosis: The roles of acylcarnitine and cell membrane–fatty acid intermediates. Vascul Pharmacol. 2016;78:17-23. doi: 10.1016/j.vph.2015.06.012
50. Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. 2017;24(1):50. doi: 10.1186/s12929-017-0357-5
51. Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget. 2016;7(45):73229-73241. doi: 10.18632/oncotarget.12302
52. Bismuth J, Lin P, Yao Q, Chen C. Ceramide: a common pathway for atherosclerosis? Atherosclerosis. 2008;196(2):497-504. doi: 10.1016/j.atherosclerosis.2007.09.018
53. Vorkas PA, Shalhoub J, Isaac G, et al. Metabolic phenotyping of atherosclerotic plaques reveals latent associations between free cholesterol and ceramide metabolism in atherogenesis. J Proteome Res. 2015;14(3):1389-1399. doi: 10.1021/pr5009898
54. Kinnunen PK, Holopainen JM. Sphingomyelinase activity of LDL: a link between atherosclerosis, ceramide, and apoptosis? Trends Cardiovasc Med. 2002;12(1):37-42. doi: 10.1016/s1050-1738(01)00143-8
55. Chaurasia B, Summers SA. Ceramides – lipotoxic inducers of metabolic disorders. Trends Endocrinol Metab. 2015;26(10):538-550. doi: 10.1016/j.tem.2015.07.006
56. Viola M, Bartolini B, Vigetti D, et al. Oxidized low density lipoprotein (LDL) affects hyaluronan synthesis in human aortic smooth muscle cells. J Biol Chem. 2013;288(41):29595-29603. doi: 10.1074/jbc.M113.508341
57. Takahashi T, Shimohata T. Vascular dysfunction induced by mercury exposure. Int J Mol Sci. 2019;20(10):2435. doi: 10.3390/ijms20102435
58. Ivanova EA, Bobryshev YV, Orekhov AN. Intimal pericytes as the second line of immune defence in atherosclerosis. World J Cardiol. 2015;7(10):583-593. doi: 10.4330/wjc.v7.i10.583
59. Lee DH, Lim JS, Song K, Boo Y, Jacobs DR Jr. Graded associations of blood lead and urinary cadmium concentrations with oxidative-stress-related markers in the U.S. population: results from the third National Health and Nutrition Examination Survey. Environ Health Perspect. 2006;114(3):350-354. doi: 10.1289/ehp.8518
60. Poręba R, Gać P, Poręba M, Andrzejak R. Environmental and occupational exposure to lead as a potential risk factor for cardiovascular disease. Environ Toxicol Pharmacol. 2011;31(2):267-277. doi: 10.1016/j.etap.2010.12.002
61. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50 Suppl(Suppl):S382-S387. doi: 10.1194/jlr.R800032-JLR200
62. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107(7):839-850. doi: 10.1161/CIRCRESAHA.110.224766
63. Watson KE, Boström K, Ravindranath R, Lam T, Norton B, Demer LL. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest. 1994;93(5):2106-2113. doi: 10.1172/JCI117205
64. Талаева Т.В., Братусь В.В. Сосудистая кальцификация: реальность и гипотезы. Медицинская газета Здоровье Украины XXI век. 2014. № 1 (32). С. 56–60.
65. Fong GH. Potential contributions of intimal and plaque hypoxia to atherosclerosis. Curr Atheroscler Rep. 2015;17(6):510. doi: 10.1007/s11883-015-0510-0
66. Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. 2014;5(8):927-946.
Унесихина Мария Сергеевна – лаборант-исследователь отдела молекулярной биологии и электронной микроскопии
ул. Попова, д. 30, г. Екатеринбург, 620014
Чемезов Алексей Игоревич – научный сотрудник отдела молекулярной биологии и электронной микроскопии
ул. Попова, д. 30, г. Екатеринбург, 620014
Сутункова Марина Петровна – д.м.н., директор
ул. Попова, д. 30, г. Екатеринбург, 620014
Унесихина М.С., Чемезов А.И., Сутункова М.П. Метаболомное профилирование при атеросклеротическом поражении сосудов и влияние тяжелых металлов на протекание заболевания (обзор литературы). Здоровье населения и среда обитания – ЗНиСО. 2022;(9):35-42. https://doi.org/10.35627/2219-5238/2022-30-9-35-42
Unesikhina M.S., Chemezov A.I., Sutunkova M.P. Metabolomic Profiling in Atherosclerotic Lesions and the Effect of Heavy Metals on the Course of Disease: A Literature Review. Public Health and Life Environment – PH&LE. 2022;(9):35-42. (In Russ.) https://doi.org/10.35627/2219-5238/2022-30-9-35-42
ЖУРНАЛ В СОЦСЕТЯХ
![]()
![]()
.png)
117105, Москва, Варшавское шоссе, 19А
ФБУЗ «Федеральный центр гигиены и эпидемиологии» Роспотребнадзора
Редакция «ЗНиСО»
E-mail: zniso@fcgie.ru






























